21 Aug 2025 | Design of Worst-Case Optimal Spaced Seeds at WABI 2025

Jens presented his paper “Design of Worst-Case Optimal Spaced Seeds” at the Wonderful Algorithms in Bioinformatics conference.




This is the group’s collection of recent and not-so-recent news.
Jens presented his paper “Design of Worst-Case Optimal Spaced Seeds” at the Wonderful Algorithms in Bioinformatics conference.

Jens and Johanna attended the Symposium on Experimental Algorithms (SEA) in Venice to present their paper “Blocked Bloom filters with choices”.
They also attended the Workshop “25 Years of Compressed Self-Indexes”.

Jens and Johanna attended the Snakemake Hackathon at the Cern in Geneva.
The Hackathon ended with the release of Snakemake 9.0.

Jens and Johanna visited the workshop Data Structures in Bioinformatics in Pisa and presented their work on probabilistic filters.
Jens gave a talk about Blocked Bloom filters with Choices. This approach improved the FPR of Blocked Bloom filters by a more balanced load per block.
Johanna gave a talk about Better Cuckoo filters, introducing a windowed memory layout to increase the fill rate of Cuckoo filters.

We had wonderful days at GCB 2024 in Bielefeld. The group was very active:
Jens presented his paper “Swiftly Identifying Strongly Unique k-Mers” at the Wonderful Algorithms in Bioinformatics conference.
We are happy to announce that Sven, Jens and Johanna are organizing a whole day tutorial at the ISMB 2024 in Montréal. We will give an introduction on Just-in-time compiled Python for bioinformatics research.

We are happy to announce that Sven, Jens and Johanna are organizing a half day workshop at the GCB 2024 in Bielefeld. We will give an introduction on Just-in-time compiled Python for bioinformatics research.

At the graduation ceremony of the SIC, Johanna was awarded the Günther Hotz Medal for an outstanding Master’s degree in Bioinformatics.
Sven, Jens and Johanna visited the workshop Data Structures in Bioinformatics in Montpellier. Johanna gave a talk about EpiSegMix, a new tool for chromatin segmentation based on epigentic marks using HMMs.
Andrea Volkamer, Sven Rahmann, and Alexey Gurevich hosted an inauguration / Christmas party at the Center of Bioinformatics.
The group visited the German Conference on Bioinformatics (GCB) in Hamburg. Sven co-organized the workshop Education in Bioinformatics. Johanna, Vu Lam and Andre presented posters about their current work. Jens gave a talk about the new Xengsort version.

Vu Lam Dang and Jens Zentgraf participated in the CPM summer school at the École normale supérieure in Paris. Vu Lam presented a poster about his work on factorization of binary matrices and the possibility of using them to calculate polygenic risk score. Jens presented a poster about the possibility to combine super-k-mers and multi-way bucketed parallel Cuckoo hashing.

From 14.06. to 16.06 Andre Holzer, Sven Rahmann, Johanna Schmitz, Johannes Schreieck and Jens Zentgraf participated in the HLA-Workshop by the Stefan Morsch Foundation. Andre gave a talk about the Opportunities and Challenges of Long-Read Sequencing.

Sven Rahmann and Jens Zentgraf visited the group of Jan Fostier in Ghent. We had an interesting exchange about search schemes, k-mers and different applications.

We visited the Stefan Morsch Foundation and had the opportunity to visit the laboratories and look at the procedure. We discussed the possibilities, difficulties and advantages of long read sequencing in HLA typing.

On 01.04 we could welcome Johanna Schmitz as a new member of the group. She has already written her master thesis entitled “Multivariate Hidden Markov Models with Flexible Distributions for Chromatin–State Discovery” in the group and is working on:
Vu Lam Dang, Sven Rahmann and Jens Zentgraf attended the Data Structures in Bioinformatics (DSB 2023) in Delft and presented parts of their work.
Sven Rahmann gave a talk about “Optimal Worst-Case Design of Gapped k-mer Masks”. Vu Lam presented joint work with Sven Rahmannm titled “Deriving polygenic risk score using non-negative matrix factorization “.

Our poster “An efficient alignment-free method for finding genetic differences between pig races from individual whole genome sequencing data” by Jens and Sven was accepted at Genome Informatics 2022, taking place September 21-23, 2022 in Hinxton, UK. Jens will present it; you can meet him there.

The 22nd Workshop on Algorithms in Bioinformatics (WABI), an annual conference with peer-reviewed proceedings published in the LIPIcs series by Dagstuhl Publishing, took place in Potsdam, Germany, this year. It was co-located with the European Symposium on Algorithms (ESA). Sven co-chaired the WABI meeting together with Christina Boucher. Jens presented his paper “Fast gapped k-mer counting with subdivided multi-way bucketed Cuckoo hash tables”.
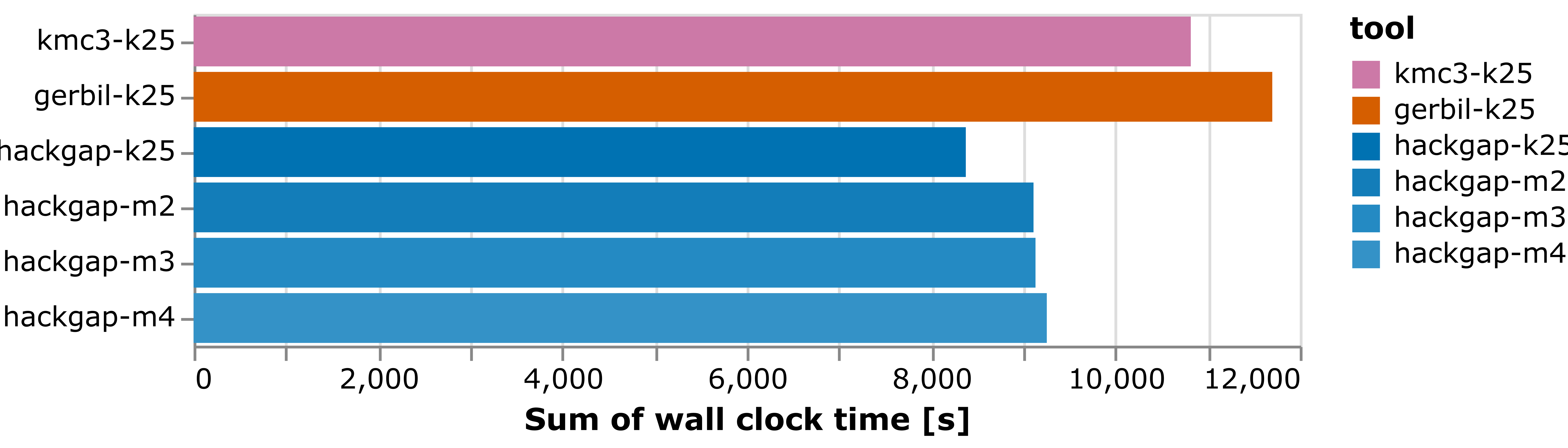
Our paper “Fast gapped k-mer counting with subdivided multi-way bucketed Cuckoo hash tables” by Jens and Sven was accepted at WABI 2022, taking place September 5-9, 2022 in Potsdam, Germany. Jens will present it; you can meet us there.
Sven, Vu Lam and Jens attended the Data Structures in Bioinformatics (DSB 2022) in Düsseldorf and presented parts of their work.
Jens Zentgraf gave a talk about joint work with Sven Rahmann, titled “Fast gapped k-mer counting with subdivided multi-way bucketed Cuckoo hash tables”.
The summer semester 2022 will offer a mixture between in-presence and online courses.
Our group offers the popular “Algorithms for Sequences Analysis” course (lectures online, may later move to an on-campus mode; tutorials both online and on-campus). More information can be found on the course website. Registration for UdS students is required to access the course materials.
Our group also offers a (Master) seminar “Algorithms for Metagenomics”. Information and registration take place via the CS Seminar website. Registration ends Tuesday, 12.04.

The Berlin-based artist Alicja Kwade had her personal genome printed out for her exhibition “In Absence” – on 314,000 DIN A4 pages of paper. To do this, she collaborated with Sven Rahmann, bioinformatics professor at Saarland University. The exhibition can be visited at the Berlinische Galerie until April 4.
The human genome consists of 3.1 billion base pairs - a number that is difficult to grasp. “Even for us bioinformaticians, this is an abstractly high number, although we work with genome data almost every day. This is because we usually only have the data as files on the computer,” says Saarbrücken bioinformatics professor Sven Rahmann.
The dimensions of the human genome can be better understood through a project by the Berlin artist Alicja Kwade. She has had her personal genome printed out on 314,000 A4 pages and is exhibiting it publicly in her exhibition “In Absence” at the Berlinische Galerie. 12,000 pages have been hung on the walls of the hall, the rest are in copper archive boxes distributed around the room. If all the pages of this genome document were laid side by side, they would stretch over a length of around 66 kilometers.
More information is available in the press release by Saarland Informatics Campus, or in an article of Saarbrücker Zeitung.
(Photograph by Frank Tschentscher)

We welcome Borja, who is finishing his PhD thesis at University of A Coruña, Spain, as a short-term guest researcher in our group. He has worked on the reconstruction of viral quasispecies, and is interested in learning about alignment-free algorithms and statistical methods in bioinformatics.

The summer term lecture “Algorithms for Sequence Analysis” by Sven Rahmann has received a Busy Beaver Award by the Student Council. This award is handed out for lectures that were well received and received very positive evaluations by students.

During the winter semester, Prof. Sven Rahmann is offering the following courses:
Lecture and tutorials in “Statistics, Probability and Applications in Bioinformatics”. More information is on the website, or directly in the SIC CMS, where you also need to register for the course with your UdS student account.
Master Seminar “Current Topics in Sequence Analysis”, for students who have previously passed the “Algorithms for Sequence Analysis course”. More information is on the website, or directly in the SIC Seminar System, where you choose your preferred seminar(s). Note that you need to go through the assignment process and cannot directly register for the seminar.

Jens Zentgraf and Sven Rahmann gave a tutorial on modern hashing methods for alignment-free (k-mer based) sequence analysis at the German Conference on Bioinformatics (GCB) 2021 online.
The slides are available online:

Saarland University Open House is taking place virtually on Satuday, June 26, 2021. All departments offer insights into their research and provide information about their study programs. We present a talk “Genome assembly as a bioinformatics puzzle with billions of pieces” for the interested public. There is also a real puzzle to download. Enjoy!
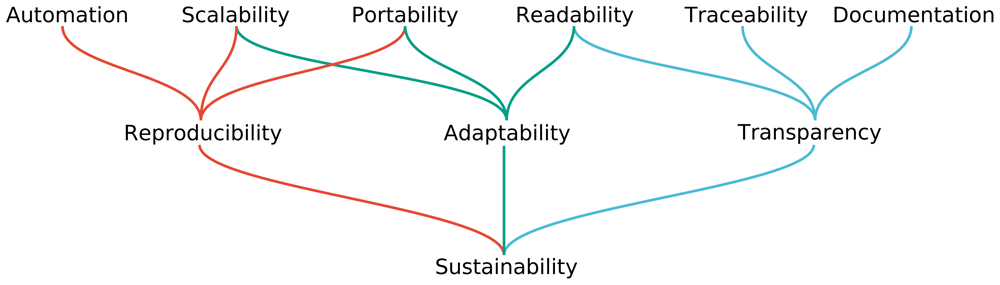
The latest version of the paper “Sustainable data analysis with Snakemake” is out.
Data analysis often entails a multitude of heterogeneous steps, from the application of various command line tools to the usage of scripting languages like R or Python for the generation of plots and tables. Here, we analyze the properties needed for a data analysis to become reproducible, adaptable, and transparent. We show how the popular workflow management system Snakemake can be used to guarantee this, and how it enables an ergonomic, combined, unified representation of all steps involved in data analysis, ranging from raw data processing, to quality control and fine-grained, interactive exploration and plotting of final results.
In this latest version, we have clarified several claims in the readability analysis. Further, we have extended the description of the scheduling to also cover running Snakemake on cluster and cloud middleware. We have extended the description of the automatic code linting and formatting provided with Snakemake. Finally, we have extended the text to cover workflow modules, a new feature of Snakemake that allows to easily compose multiple external pipelines together, while being able to extend and modify them on the fly.
During this semester, the “Algorithms for Sequences Analysis” course is offered by Sven Rahmann.
More information can be found on the course website. Registration for UdS students is required via the Course Management System of Saarland Informatics Campus.
We present GAMIBHEAR, a tool for accurate haplotype reconstruction from GAM data. GAMIBHEAR aggregates allelic co-observ ation frequencies from GAM data and employs a GAM-specific probabilistic model of haplotype capture to optimize phasing accuracy.
“GAMIBHEAR” is available as an R package under the open-source GPL-2 license. The paper has been published in the Bioinformatics journal. “GAMIBHEAR: whole-genome haplotype reconstruction from Genome Architecture Mapping data”
Genome Architecture Mapping (GAM) was recently introduced as a digestion- and ligation-free method to detect chromatin conformation.GAM’s ability to capture both inter- and intra-chromosomal contacts from low amounts of input data makes it particularly well suited for allele-specific analyses in a clinical setting. Allele-specific analyses are powerful tools to investigate the effects of genetic variants on many cellular phenotypes including chromatin conformation, but require the haplotypes of the individuals under study to be known a priori. So far, however, no algorithm exists for haplotype reconstruction and phasing of genetic variants from GAM data, hindering the allele-specific analysis of chromatin contact points in non-model organisms or individuals with unknown haplotypes.

As of today, Prof. Dr. Sven Rahmann heads the new Algorithmic Bioinformatics group at Saarland University. The group belongs to both the Mathematics and Informatics Faculty (“MI”) and the Center for Bioinformatics (ZBI) at Saarland University and is part of Saarland Informatics Campus (SIC).
The research focus will gradually shift more towards algorithm development and methodological research. We look forward towards new exciting projects and collaborations, and we are also happy to continue and conclude ongoing projects at University Alliance Ruhr. This website will remain the website of the lab, and the content will be gradually adapted towards the new profile.
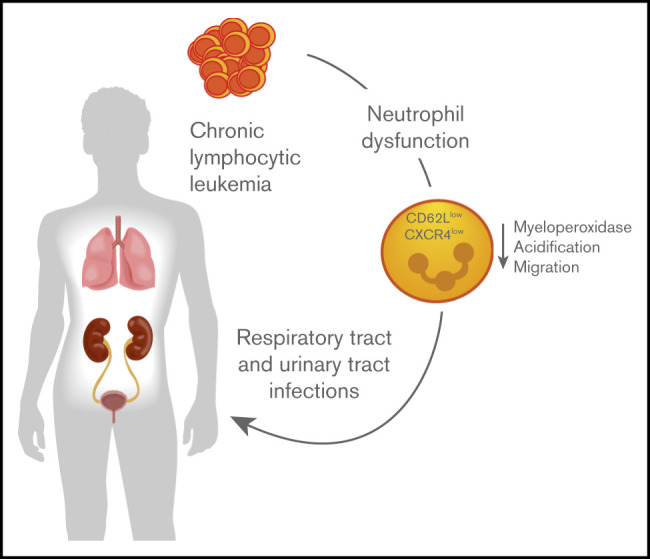
Our paper Proteomic and bioinformatic profiling of neutrophils in CLL reveals functional defects that predispose to bacterial infections is online.
Patients with chronic lymphocytic leukemia (CLL) typically suffer from frequent and severe bacterial infections. Although it is well known that neutrophils are critical innate immune cells facilitating the early defense, the underlying phenotypical and functional changes in neutrophils during CLL remain largely elusive. Using a murine adoptive transfer model of CLL, we demonstrate aggravated bacterial burden in CLL-bearing mice upon a urinary tract infection with uropathogenic Escherichia coli. Bioinformatic analyses of the neutrophil proteome revealed increased expression of proteins associated with interferon signaling and decreased protein expression associated with granule composition and neutrophil migration. Functional experiments validated these findings by showing reduced levels of myeloperoxidase and acidification of neutrophil granules after ex vivo phagocytosis of bacteria. Pathway enrichment analysis indicated decreased expression of molecules critical for neutrophil recruitment, and migration of neutrophils into the infected urinary bladder was significantly reduced. These altered migratory properties of neutrophils were also associated with reduced expression of CD62L and CXCR4 and correlated with an increased incidence of infections in patients with CLL.
In conclusion, this study describes a molecular signature of neutrophils through proteomic, bioinformatic, and functional analyses that are linked to a reduced migratory ability, potentially leading to increased bacterial infections in patients with CLL.
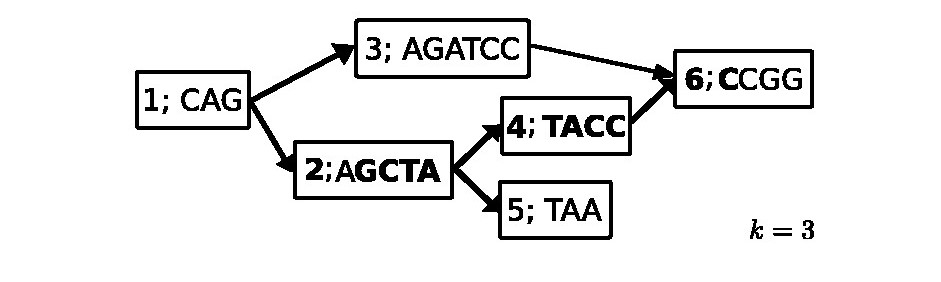
Our paper Detecting high-scoring local alignments in pangenome graphs is about a new heuristic method to find maximum scoring local alignments of a DNA query sequence to a pangenome represented as a compacted colored de Bruijn graph.
Our approach additionally allows a comparison of similarity among sequences within the pangenome. We show that local alignment scores follow an exponential-tail distribution similar to BLAST scores, and we discuss how to estimate its parameters to separate local alignments representing sequence homology from spurious findings. An implementation of our method is presented, and its performance and usability are shown. Our approach scales sublinearly in running time and memory usage with respect to the number of genomes under consideration. This is an advantage over classical methods that do not make use of sequence similarity within the pangenome.
PLAST builds a compacted, colored de Bruijn graph from given input genomes using the API of Bifrost. Apart from the requirements of Bifrost (c++ and cmake), there are no further strict dependencies. The source code and test data is available here: PLAST

Fully-funded 3-year position (TV-L E13, 100%) for a doctoral student (m/f/d) in our Research Training Group WisPerMed (“Knowledge and data based personalization of medicine at the point of care”) in a research project on genome-wide variant data and its interpretation. Use and learn Snakemake, Python, Rust, … Develop software that will make a difference in clinical practice with a focus on reproducibility and code quality. Help future doctors to access patient-related information quickly and in context. Work with Johannes Köster and Sven Rahmann. Please take a look at this poster and apply immediately via WisPerMed or the German version.
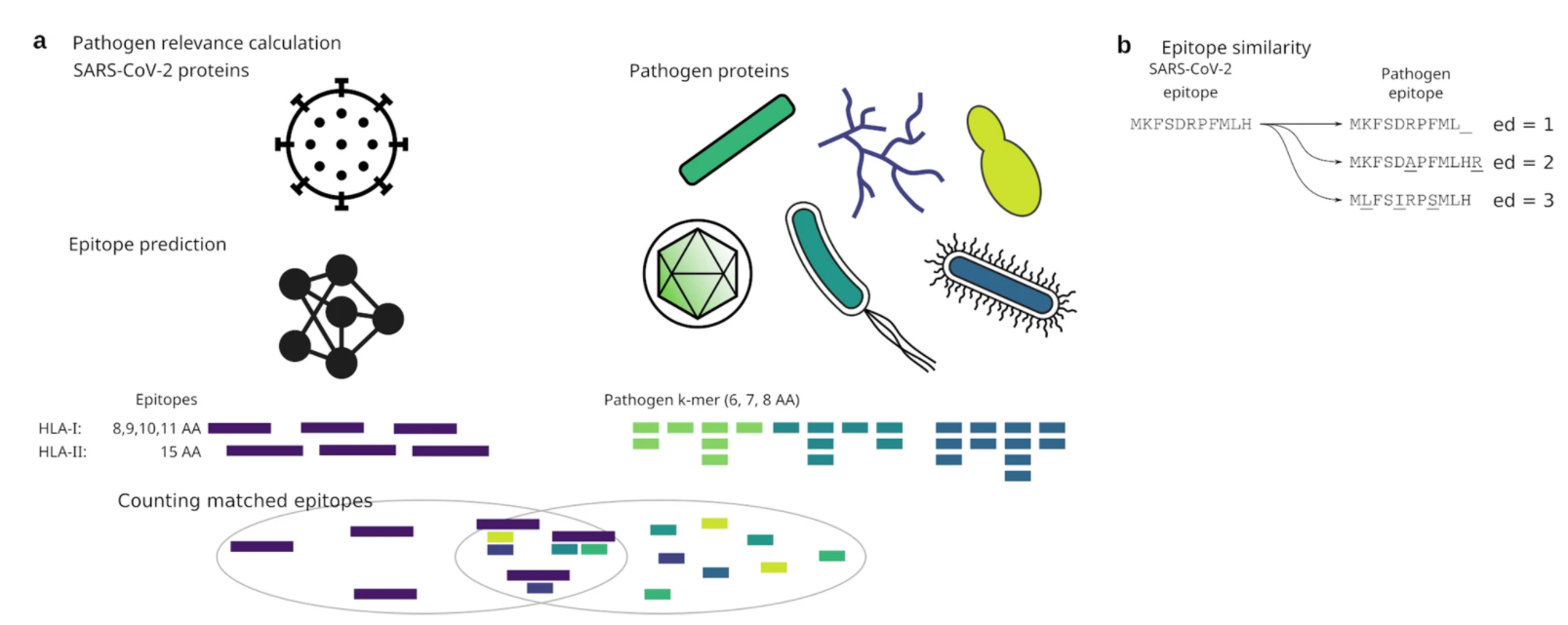
Our findings on SARS-CoV-2 viral proteins is published in Scientific Reports-Nature. The paper Epitope similarity cannot explain the pre-formed T cell immunity towards structural SARS-CoV-2 proteins is online.
The current pandemic is caused by the SARS-CoV-2 virus and large progress in understanding the pathology of the virus has been made since its emergence in late 2019. Several reports indicate short lasting immunity against endemic coronaviruses, which contrasts studies showing that biobanked venous blood contains T cells reactive to SARS-CoV-2 S-protein even before the outbreak in Wuhan. This suggests a preformed T cell memory towards structural proteins in individuals not exposed to SARS-CoV-2. Given the similarity of SARS-CoV-2 to other members of the Coronaviridae family, the endemic coronaviruses appear likely candidates to generate this T cell memory. However, given the apparent poor immunological memory created by the endemic coronaviruses, immunity against other common pathogens might offer an alternative explanation. Here, we utilize a combination of epitope prediction and similarity to common human pathogens to identify potential sources of the SARS-CoV-2 T cell memory. Although beta-coronaviruses are the most likely candidates to explain the pre-existing SARS-CoV-2 reactive T cells in uninfected individuals, the SARS-CoV-2 epitopes with the highest similarity to those from beta-coronaviruses are confined to replication associated proteins-not the host interacting S-protein.
Thus, our study suggests that the observed SARS-CoV-2 pre-formed immunity to structural proteins is not driven by near-identical epitopes.
Our article entitled Machine learning reveals a PD-L1–independent prediction of response to immunotherapy of non-small cell lung cancer by gene expression context is published in European Journal of Cancer and now available online.
We set out to apply context-sensitive feature selection and machine learning approaches on expression profiles of immune-related genes in diagnostic biopsies of patients with stage IV NSCLC.We applied supervised machine learning methods for feature selection and generation of predictive models.Feature selection and model creation were based on a training cohort of 55 patients with recurrent NSCLC treated with PD-1/PD-L1 antibody therapy. Resulting models identified patients with superior outcomes to immunotherapy, as validated in two subsequently recruited, separate patient cohorts (n = 67, hazard ratio = 0.46, p = 0.035). The predictive information obtained from these models was orthogonal to PD-L1 expression as per immunohistochemistry: Selecting by PD-L1 positivity at immunohistochemistry plus model prediction identified patients with highly favourable outcomes.Visualisation of the models revealed the predictive superiority of the entire 7-gene context over any single gene.
Using context-sensitive assays and bioinformatics capturing the tumour immune context allows precise prediction of response to PD-1/PD-L1-directed immunotherapy in NSCLC.
Our paper A germ cell-specific ageing pattern in otherwise healthy men is online.
Life-long sperm production leads to the assumption that male fecundity remains unchanged throughout life. However, recently it was shown that paternal age has profound consequences for male fertility and offspring health. Paternal age effects are caused by an accumulation of germ cell mutations over time, causing severe congenital diseases. Apart from these well-described cases, molecular patterns of ageing in germ cells and their impact on DNA integrity have not been studied in detail.
In this study, we aimed to assess the effects of ‘pure’ ageing on male reproductive health and germ cell quality. We assembled a cohort of 198 healthy men (18–84 years) for which end points such as semen and hormone profiles, sexual health and well-being, and sperm DNA parameters were evaluated. Sperm production and hormonal profiles were maintained at physiological levels over a period of six decades. In contrast, we identified a germ cell-specific ageing pattern characterized by a steady increase of telomere length in sperm and a sharp increase in sperm DNA instability, particularly after the sixth decade. Importantly, we found sperm DNA methylation changes in 236 regions, mostly nearby genes associated with neuronal development. By in silico analysis, we found that 10 of these regions are located in loci which can potentially escape the first wave of genome-wide demethylation after fertilization.
In conclusion, human male germ cells present a unique germline-specific ageing process, which likely results in diminished fecundity in elderly men and poorer health prognosis for their offspring.
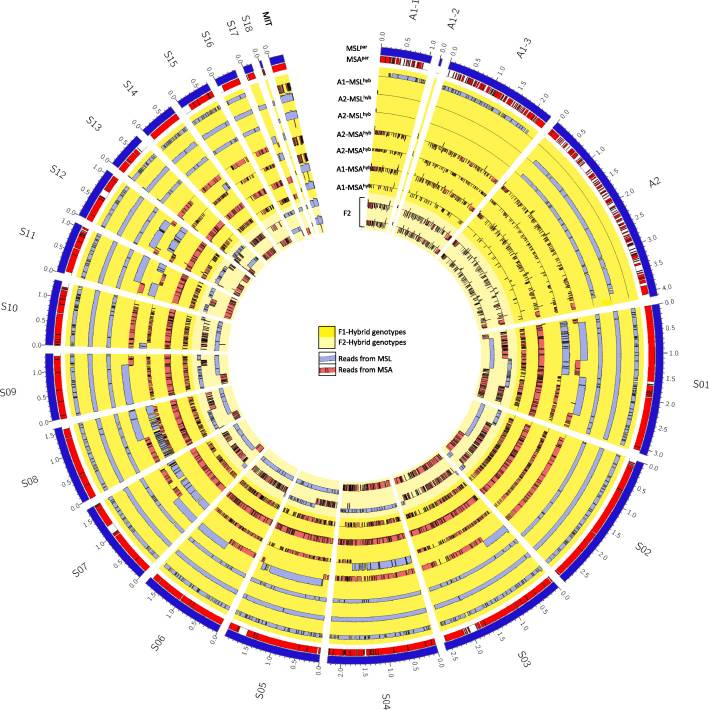
Our article Meiotic recombination in the offspring of Microbotryum hybrids and its impact on pathogenicity is published in BMC Evolutionary Biology.
Here, we performed experimental crosses between the two pathogenic Microbotryum species, M. lychnidis-dioicae and M. silenes-acaulis that are specialized to different hosts. The resulting offspring were analyzed on phenotypic and genomic levels to describe genomic characteristics of hybrid offspring and genetic factors likely involved in host-specialization.
Genomic analyses of interspecific fungal hybrids revealed that individuals were most viable if the majority of loci were inherited from one species. Interestingly, species-specific loci were strictly controlled by the species’ origin of the mating type locus. Moreover we detected signs of crossing over and chromosome duplications in the genomes of the analyzed hybrids. In Microbotryum, mitochondrial DNA was found to be uniparentally inherited from the a2 mating type. Genome comparison revealed that most gene families are shared and the majority of genes are conserved between the two species, indicating very similar biological features, including infection and pathogenicity processes. Moreover, we detected 211 candidate genes that were retained under host-driven selection of backcrossed lines. These genes and might therefore either play a crucial role in host specialization or be linked to genes that are essential for specialization.
This study manifests genetic factors of host specialization that are required for successful biotrophic infection of the post-zygotic stage, but also demonstrates the strong influence of intra-genomic conflicts or instabilities on the viability of hybrids in the haploid host-independent stage.
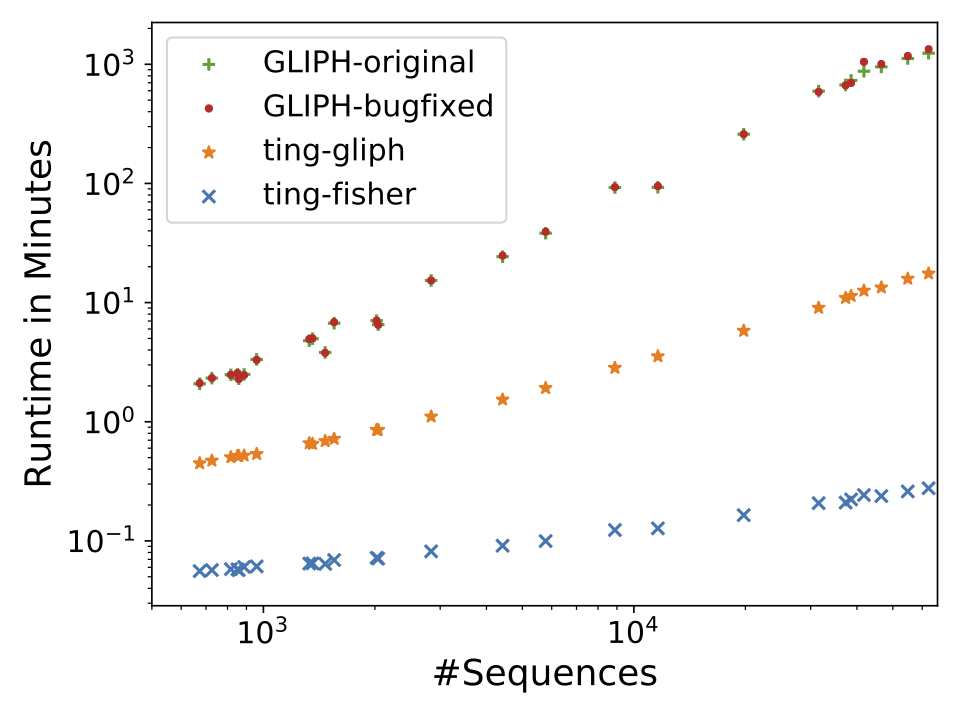
Our preprint “Rapid T cell receptor interaction grouping with ting” is online. Clustering of antigen-specific T cell receptor repertoire (TCRR) sequences remains challenging. While established tools like gliph aim to solve this problem they suffer from serveral shortcommings, including bad performance on huge repertoires, non-determinism, potential loss of significant antigen-specific or inclusion of too many unspecific sequences. “ting” solves these issues by applying an efficient algorithm for identifying antigen-specific k-mers based on Fisher’s Exact Test. This allows fast processing of large scale repertoires and an improved differentiation between naive and specific TCR3b sequences.
The full paper has been submitted for review.
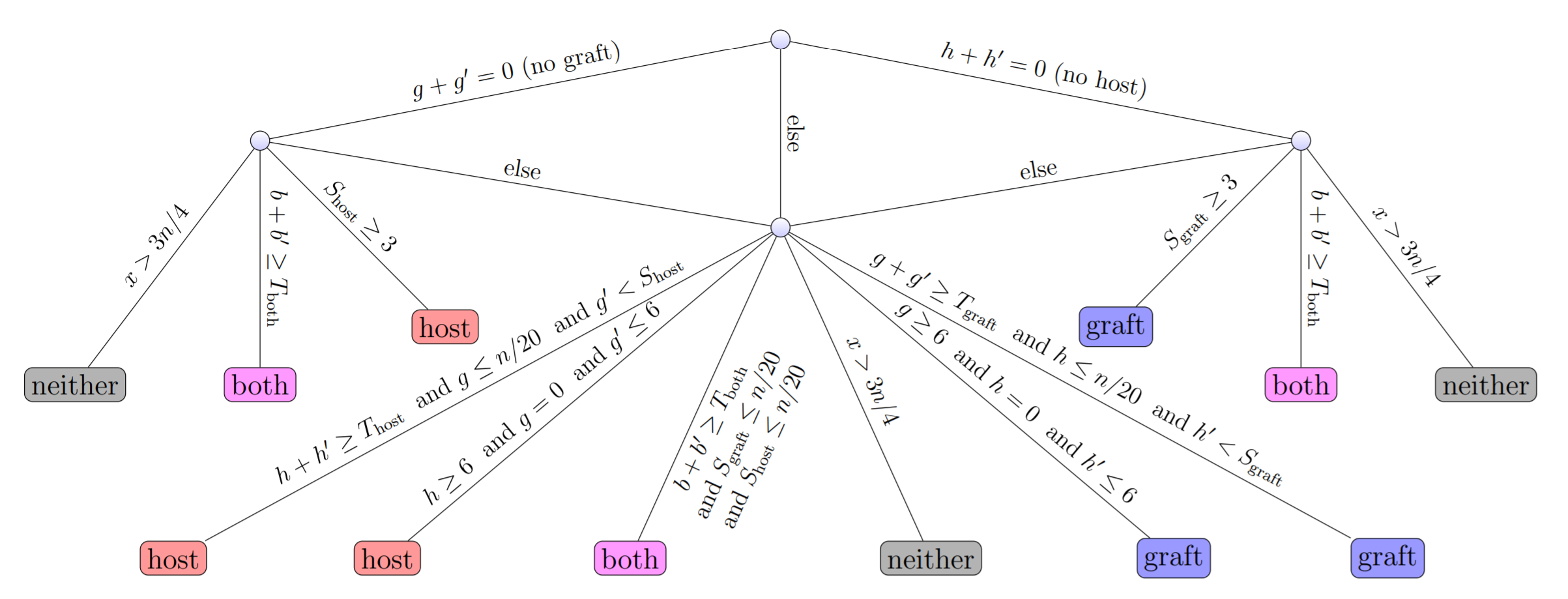
Finally, our preprint “Fast lightweight accurate xenograft sorting” is online. Xenograft sorting classifies the (paired-end or single-end) reads of a xenograft sample according to species of origin. A typical application concnerns sequenced samples from patient-derived xenografts (PDX; tumors extracted from human patients and implanted into mice), where the reads have to be classified into human reads and mouse reads (and, possibly, reads that could originate from both species, reads from neither species, and ambiguous reads). We have developed an alignment-free approach based on 3-way bucketed Cuckoo hashing. Our tool “xengsort” is faster by a factor of 4 than existing alignment-free tools on typical PDX datasets.
A poster about this work will be presented at ISMB HiTSeq. The full paper has been submitted for review.
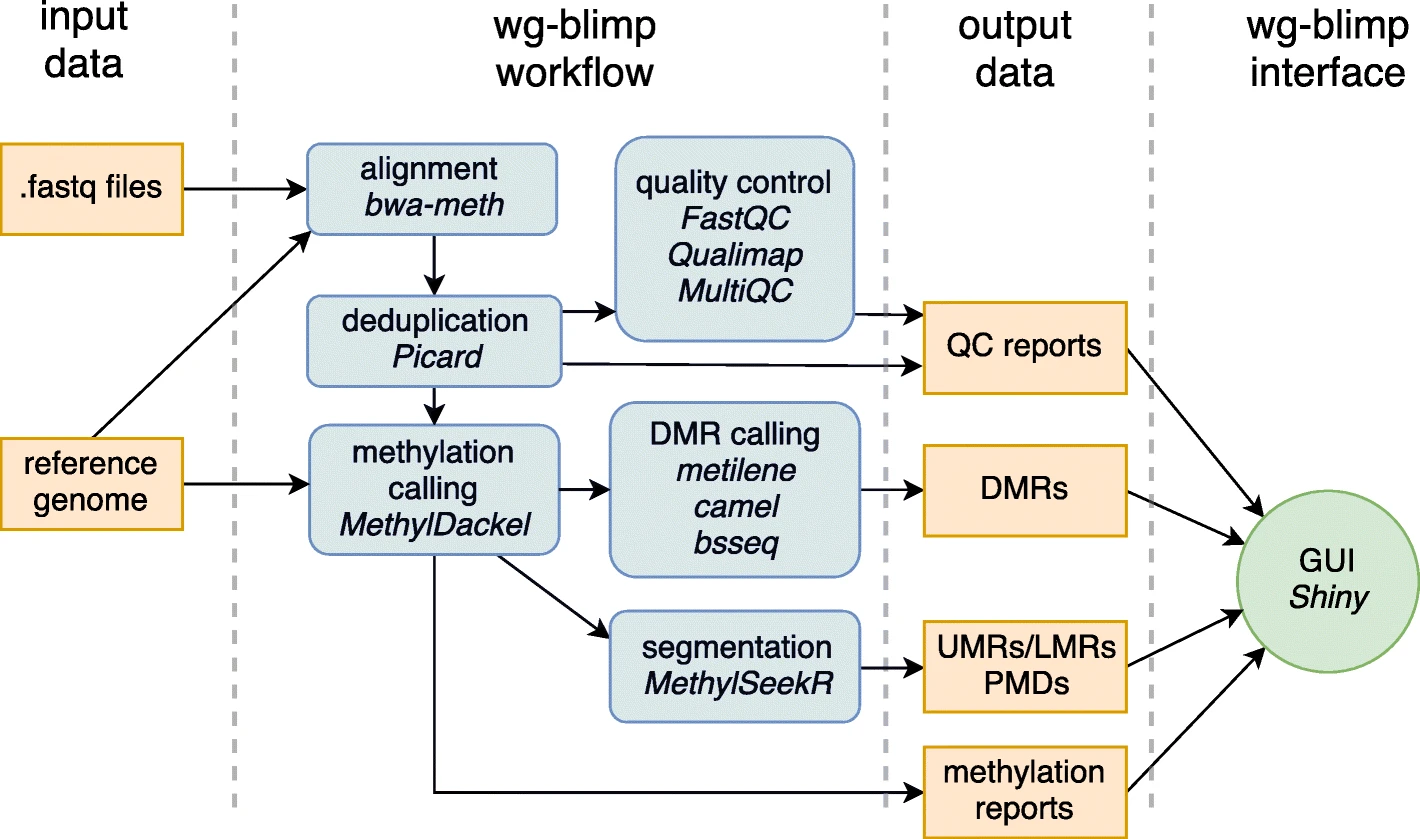
Our article “wg-blimp: an end-to-end analysis pipeline for whole genome bisulfite sequencing data”has been published in BMC Bioinformatics journal.
We developed wg-blimp (whole genome bisulfite sequencing methylation analysis pipeline) as an end-to-end pipeline to ease whole genome bisulfite sequencing data analysis. It integrates established algorithms for alignment, quality control, methylation calling, detection of differentially methylated regions, and methylome segmentation, requiring only a reference genome and raw sequencing data as input. We improve on previous pipelines by providing a more comprehensive analysis workflow as well as an interactive user interface.
We demonstrated its applicability by analysing multiple publicly available datasets. Thus, wg-blimp is a relevant alternative to previous analysis pipelines and may facilitate future epigenetic research.
Elias’ and Sven’s paper “Engineering Fused Lasso Solvers on Trees” was accepted at the 18th Symposium on Experimental Algorithms (SEA 2020), which will be held as an online conference from June 16 till June 18. Our paper presents two practically efficient algorithms for solving fused lasso problems on tree graphs with general weights for nodes and edges, even zero weights, which other algorithms cannot easily handle.
We hope to see you at SEA’20 for our presentation. Also, the TreeLas software is avaliable on our Software page.
Aufgrund der Beschlüsse der Landesregierung NRW und der einzelnen Universitäten in NRW findet ab 20.04. online-Lehre statt. Das betrifft insbesondere “Algorithmen auf Sequenzen” von Prof. Rahmann und die Vorbesprechung zum Seminar “Aktuelle Themen der Bioinformatik”. Ferner werden ausstehende Prüfungen (z.B. zu “Algorithmische Bioinformatik”) online durchgeführt.

Two researchers from the Genome Informatics group attended the SIGOPT 2020 meeting in Dortmund and presented their work on continous and combinatorial optimization problems in bioinformatics.
Elias presented his fast solver for the fused lasso problem on tree graphs (joint work with Sven).
Jens gave a talk about joint work with Henning and Sven, titled “Cost-optimal assignment of elements in genome-scale multi-way bucketed Cuckoo hash tables”.
The bioinformatics session on Friday further included talks by David Blumenthal (Munich) on median graphs and by Sven Schrinner (Düsseldorf) on polyploid phasing.

Researchers from the Genome Informatics group attended the Data Structures in Bioinformatics (DSB 2020) in Rennes and presented their work.
Jens Zentgraf gave a talk about joint work with Henning Timm and Sven Rahmann, titled “Cost-optimal assignment of elements in genome-scale multi-way bucketed Cuckoo hash tables”.
Sven Rahmann presented joint work with Jens Zentgraf on “Faster xenograft sorting with 3-way bucketed Cuckoo hash tables”.
Even though it was raining in Rennes and Air France wasn’t able to fly from Düsseldorf to Rennes in under 9 hours, the meeting was a lot of fun and showcased many interesting new results.

Dr. Fabian Kilpert is joining the Genome Informatics group in December 2019. Together with the new director of the Institute of Human Genetics, he is coming to Essen from Lübeck in Schleswig-Holstein, where he previously worked at the Institute of Neurogenetics. Welcome, Fabian!

The third annual meeting of the INCEPTION program took place at Institut Pasteur in Paris on November 18 and 19. A distinguishing feature of this program is the tight collaboration between the life sciences and social sciences. As a member of the scientific advisory board, Sven gave a talk about “reproducible genome-wide k-mer association studies”.
Our paper “Cost-optimal assignment of elements in genome-scale multi-way bucketed Cuckoo hash tables” by Jens, Henning and Sven was accepted at ALENEX 2020, taking place January 5-6, 2020 at Salt Lake City, Utah, USA. Jens will present it; you can meet us there.
In the winter semester 2019/20, the Genome Informatics group offers the Master specialization course “Algorithmic Bioinformatics” at TU Dortmund.

Bianca Stöcker presents her work “Protein Complex Similarity Based on Weisfeiler-Lehman Labeling” at the “Similarity Search and Applications” (SISAP) conference at Newark, New Jersey, USA.
Protein complexes consist of several protein chains. The similarity between two complexes can be measured by comparing the sets or multisets of their protein chains, but it is conceivable that two complexes consist of the same (multi)set of chains, but has a different topology, i.e. different physical interactions between the chains. This should be reflected in the similarity measure. Our work defines such a similarity measure that incorporates topological information but is almost as efficiently computable than set-based measures.
This publication resulted from a collaboration within the DFG-funded Collaborative Research Center SFB 876 between members of projects A6 (Resource-efficient graph mining) and C1 (Feature selection in high dimensional data for risk prognosis in oncology). This work is also part of our long-running project on protein hypernetworks and constraints between protein interactions.
As of today, Prof. Dr. Frank Kaiser is the new director of the Insititute of Human Genetics at the University of Duisburg-Essen. Welcome, Frank! We look forward to many exciting new collaborations. The former director, Prof. Dr. Bernhard Horsthemke, will continue to work as a senior professor at the institute, and we are happy to continue our joint projects.
At a computational pan-genomics workshop at the Center for Interdisciplinary Research (ZiF) at Bielefeld University, Sven presented joint work with Jens Quedenfeld on the analysis of min-hashing for variant tolerant read mapping. Genome Informatics alumnus Tobias Marschall gave a talk about “Rapid bit parallel sequence to graph alignment”. The workshop was organized by Jens Stoye and Alexander Schönhuth.

The Schering Sitftung has awarded the Friedmund Neumann Prize 2019 to Johannes Köster. Congratulations, Johannes!
Image credits: Friedmund Neumann Prize 2019 of the Schering Stiftung. Award ceremony, 24.09.2019 in Berlin. Photo: Julia Zimmermann, © Schering Stiftung. Persons (left-to-right): Prof. Dr. Sven Rahmann (nominator), Dr. Johannes Köster (awardee), Prof. Dr. Dr. h. c. Stefan H. E. Kaufmann (chairman of the Foundation Concil).
There are more pictures from the award ceremony available at the Schering Stiftung website.

Several researchers from the Genome Informatics group attended the German Conference on Bioinformatics (GCB) 2019 in Heidelberg and presented their work:

Jens Zentgraf and Sven Rahmann gave a tutorial on foundations of alignment-free sequence analysis: k-mer hashing at the German Conference on Bioinformatics (GCB) 2019 in Heidelberg.
Over 20 participants attended the tutorial. It was a lot of fun; we may do it again soon.

The Deutsche Forschungsgemeinschaft (DFG) funds the Collaborative Research Center SFB 876 “Providing Information by Resource-Constrained Data Analysis” for additional four years (2019, 2020, 2021, 2022). The Chair of Genome Informatics, in particular Till Hartmann and Sven Rahmann, will contribute towards project C1 (“Feature selection in high dimensional data for risk prognosis in oncology”) and work on efficient methods for dimensionality reduction and resource-efficicent analysis methods for nanopore sequencing data.

The article “Analysis of min-hashing for variant tolerant DNA read mapping” by Jens Quedenfeld (now at TU Munich) and Sven Rahmann has received the Best Paper Award at the Workshop of Algorithms in Bioinformatics (WABI) 2017, held in Cambridge, MA, USA, August 20-23, 2017.
The authors consider an important question, as DNA read mapping has become a ubiquitous task in bioinformatics. New technologies provide ever longer DNA reads (several thousand basepairs), although at comparatively high error rates (up to 15%), and the reference genome is increasingly not considered as a simple string over ACGT anymore, but as a complex object containing known genetic variants in the population. Conventional indexes based on exact seed matches, in particular the suffix array based FM index, struggle with these changing conditions, so other methods are being considered, and one such alternative is locality sensitive hashing. Here we examine the question whether including single nucleotide polymorphisms (SNPs) in a min-hashing index is beneficial. The answer depends on the population frequency of the SNP, and we analyze several models (from simple to complex) that provide precise answers to this question under various assumptions. Our results also provide sensitivity and specificity values for min-hashing based read mappers and may be used to understand dependencies between the parameters of such methods. This article may provide a theoretical foundation for a new generation of read mappers.
The article can be freely accessed in the WABI conference proceedings (Proceedings of the 17th International Workshop on Algorithms in Bioinformatics (WABI 2017), Russell Schwartz and Knut Reinert (Eds.), LIPICS Vol. 88).
This work is part of subproject C1 of the collaborative research center SFB 876.
Algorithmic Bioinformatics, SIC, Saarland University | Privacy notice | Legal notice