



Feature selection in high dimensional data for risk prognosis in oncology
Investigators: Bianca Stöcker, Till Hartmann, Johannes Köster & Sven Rahmann
Collaborators: Collaborative Research Center SFB 876
Funding: DFG SFB 876/3, project C1
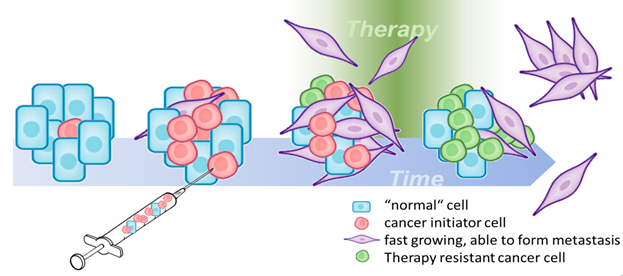
Recent advances in molecular biotechnologies have fundamentally changed how cancer patients are diagnosed and treated. The development of targeted therapies has increased patients’ life expectancy and quality of life with the majority of cancer types. However, predicting treatment efficacy and selecting the optimal personalised therapy for each patient remains a challenge for clinicians. Mainly, the development of resistance to therapy and intratumoural heterogeneity limit successful long-term remissions and cures. The early prediction of therapy resistance or relapse is thus deemed crucial for further improving therapy outcome. Identification of features termed biomarkers, which are derived from patient samples by high-throughput analyses, is an important means to achieve this goal. Project C1 builds and optimises models for clinically relevant decisions in oncology by selecting features from high-dimensional feature spaces, extracted from raw data created on different molecular platforms.
In the past, highly parallel (“next generation”) DNA sequencing technology allowed researchers with access to specialised sequencing core facilities to discover tumour-specific mutations. As DNA sequencing capacity continues to increase and costs continue to drop faster than computational capacity and storage can keep up, new algorithmic paradigms are needed for the analysis of very large genomic data sets. In project C1, we investigate new algorithms to extract relevant features for biomarker discovery from whole-genome data sets in the 10–100 terabyte range on commodity hardware by streaming the sequence data and filtering for features of interest using novel string hashing methods.
Recent developments in nanopore sequencing are democratising DNA sequencing and genome analysis. The new nanopore sequencers are of size comparable to a USB stick, are inexpensive, and can be used without specialised lab equipment. While nanopore sequencing offers lower throughput and higher error rates than established technologies at the moment, it has the potential to turn DNA sequencing and subsequently genomic analysis into a commodity. In oncology, the vision is that nanopore sequencing, together with non-invasive patient monitoring techniques such as “liquid biopsies” drawn from blood or urine, will allow for detection of small amounts of circulating tumour DNAs, allowing an accurate assessment of patient risk and therapy options. In principle, such an assessment would be possible anywhere at any time, given standard equipment of moderate costs, i.e., the sequencer and a laptop or embedded system.
For this vision to become a reality, several data analysis challenges must be overcome: In addition to the constraints imposed by small sample size n compared to the high dimensionality p of the feature space (n « p problem), the cyber-physical systems for nanopore sequencing create novel resource constraints: The raw data generated by this new technology is a large-volume high-frequency signal of ion currents, which is difficult to translate directly into a DNA sequence. Therefore, to identify tumour “fingerprints” or biomarkers based on tracing tumour-derived nucleic acids, either better methods for DNA base calling from ion currents are needed, or a different representation of the tumour fingerprints has to be considered, such as features in signal space. We will follow both avenues in parallel and in particular consider novel features derived from a discretised compressed ion current signal space.
Algorithmic Bioinformatics, SIC, Saarland University | Privacy notice | Legal notice