



Metatranscriptomics
Investigators: Daniela Beisser & Sven Rahmann
Collaborators: Jens Boenigk (biodiversity chair), Essen
Funding: internal
Software: TaxMapper
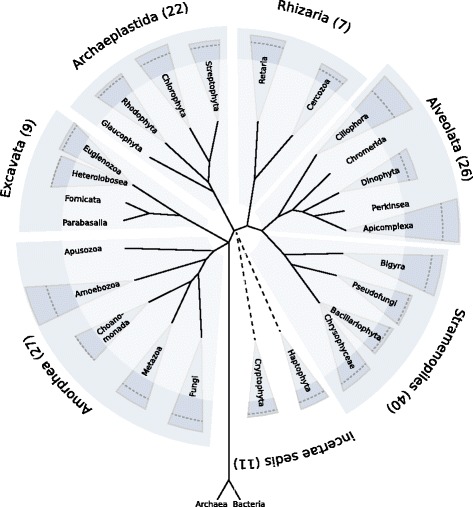
Next generation sequencing technologies are increasingly applied to analyse complex ecosystems by mRNA sequencing of whole communities. In principle, each sequenced mRNA allows identifying the underlying species as well as a functional annotation. While the functional information is sufficiently covered by databases such as Uniprot and NCBI, the approach is currently limited by incomplete taxonomic references.
For an accurate assignment of taxonomic groups to environmental metatranscriptomic reads we build a custom database that comprises all major eukaryotic groups and a stand-alone tool to assign reads with a low false discovery rate. The custom database includes peptide sequences translated from transcriptomes of all relevant eukaryotic taxonomic groups, in total 146 species. We do not attempt to assign sequence reads on species or genera level, but to higher taxonomic groups. The biggest problem is the misassignment of sequences to incorrect species, we therefore perform rigorous filtering, in which we evaluate the distance between the best hit and next hit in another taxonomic group.
The developed tool TaxMapper, available at bitbucket and bioconda, is built in a modular way to be applicable stepwise with user-set parameters or as a complete easy-to-use analysis with standard parameters starting from a RAPsearch mapping file to a visualization of community composition. Additionally, we develop a reliable workflow for microeukaryotic metatranscriptome analysis. Written as a rule-based Snakemake workflow, it unites all major bioinformatic steps:
- preprocessing of raw reads,
- functional and taxonomic assignment with TaxMapper,
- and statistical analyses.
The setup can be adjusted to any environmental sample.
Algorithmic Bioinformatics, SIC, Saarland University | Privacy notice | Legal notice