



Multi-way bucketed Cuckoo hashing for alignment-free sequence analysis
Investigators: Jens Zentgraf & Sven Rahmann
Funding: internal
Software: xengsort
In recent years, alignment free sequence analysis methods have gained importance, due to their superior speed at equivalent results in comparison to traditional mapping- and alignment-based methods. Recently, methods have emerged that are able to index very large collections of sequenced DNA samples (e.g. any genome ever sequenced).
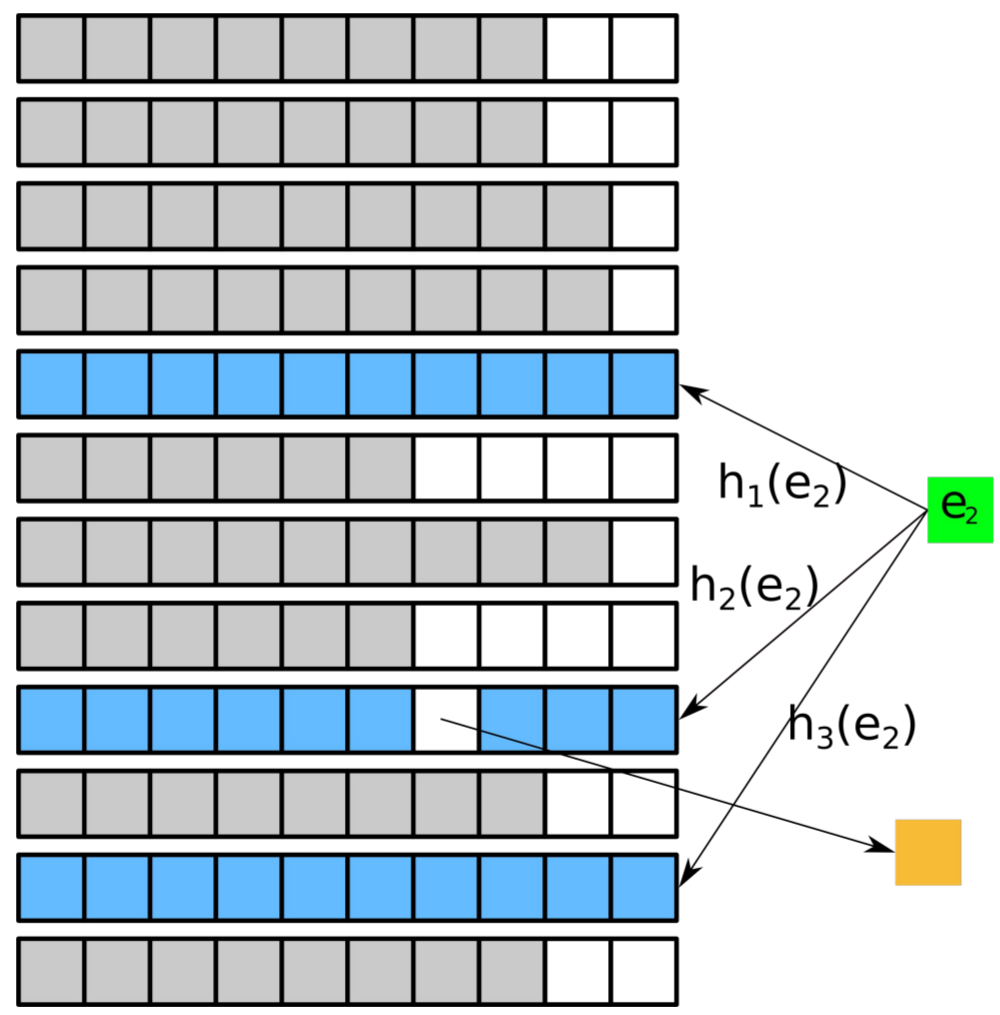
The basis of each alignment-free method is a so called k-mer dictionary (or key-value-store) hat associates a value (e.g., a transcript ID, chromosome number, species ID or counter) to each DNA substring of length k (from a genome or a sequenced sample). Almost always, such a dictionary is implemented via hashing. Ideally, considering that billions of k-mers have to be processed, such a hash table is both small and fast. It is both a science and an art to design fast and small hash tables for a given task.
In this project, we engineer multi-way bucketed Cuckoo hash tables using dynamic compilation at runtime and several other tricks of the trade to obtain so far unseen ultra-fast retrieval times. We also keep the memory requirements low by resorting to high fill rates (load factors) and techniques like quotienting.
Applications
We apply the engineered hash tables to several applications for alignment-free (k-mer based) sequence analysis, for example:
- xenograft sorting, i.e., classifying reads from a mixed sample containing graft tumor and host reads
- general-purpose k-mer counting
- metagenomic read classification
Tutorial on modern hashing at GCB 2021
Jens Zentgraf and Sven Rahmann gave a tutorial on modern hashing methods for alignment-free (k-mer based) sequence analysis at the German Conference on Bioinformatics (GCB) 2021 online.
The slides are available online:
- Introduction: k-mers and alignment-free methods
- Hashing: Hash functions and collision resolution strategies
- Multi-way bucketed Cuckoo hashing
- Performance engineering
Publications
- Jens Zentgraf & Sven Rahmann. “Fast lightweight accurate xenograft sorting”. Algorithms for molecular biology 16(1):2; Apr 2021.
- Jens Zentgraf, Henning Timm & Sven Rahmann: Cost-optimal assignment of elements in genome-scale multi-way bucketed Cuckoo hash tables. ALENEX 2020: 186-198. SIAM.
Algorithmic Bioinformatics, SIC, Saarland University | Privacy notice | Legal notice