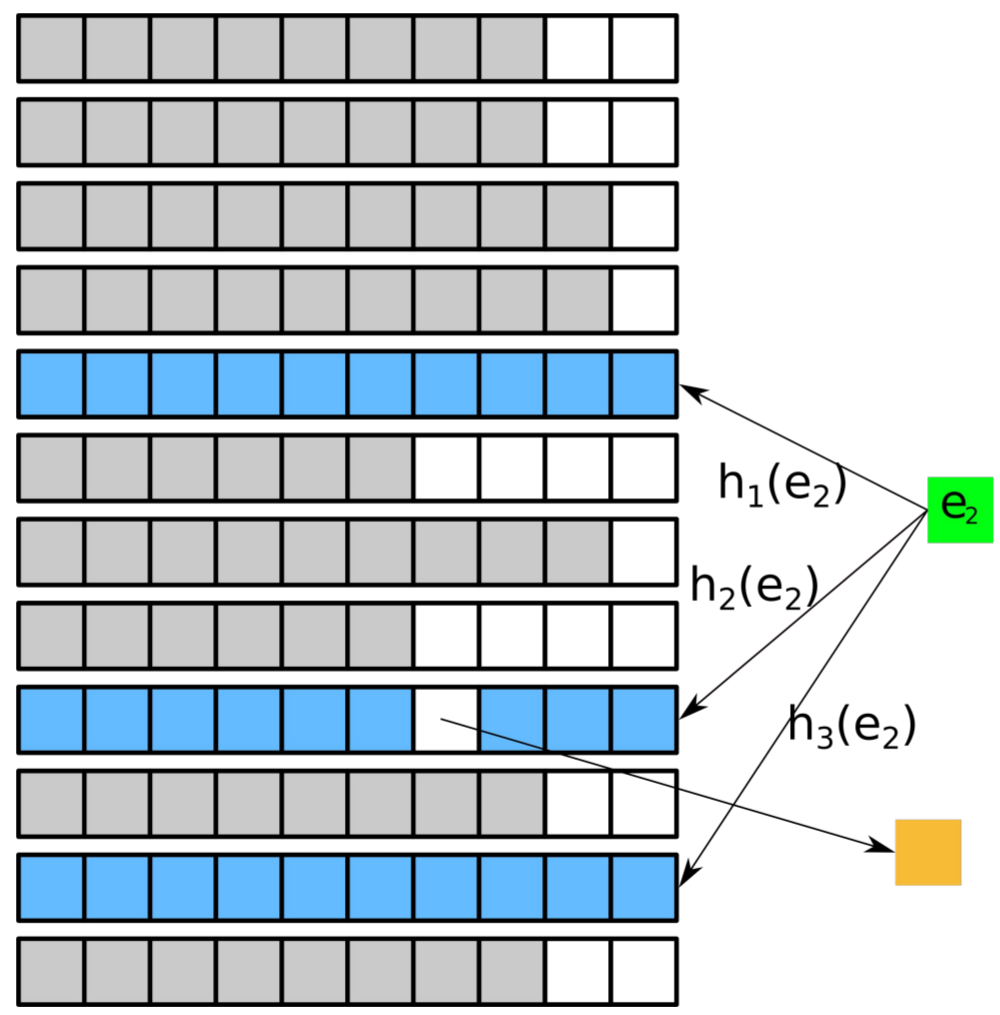
Design of gapped k-mer seeds
 Investigators: Kamalika Ray & Sven Rahmann
Investigators: Kamalika Ray & Sven Rahmann
Funding: internal
Summary: We develop methods for optimizing gapped k-mer seeds with regard to beneficial properties, such as tolerance towards mismatches in a sequenced read.
(Details)
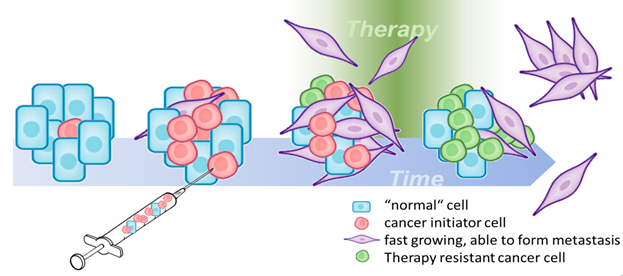
Feature selection in high dimensional data for risk prognosis in oncology
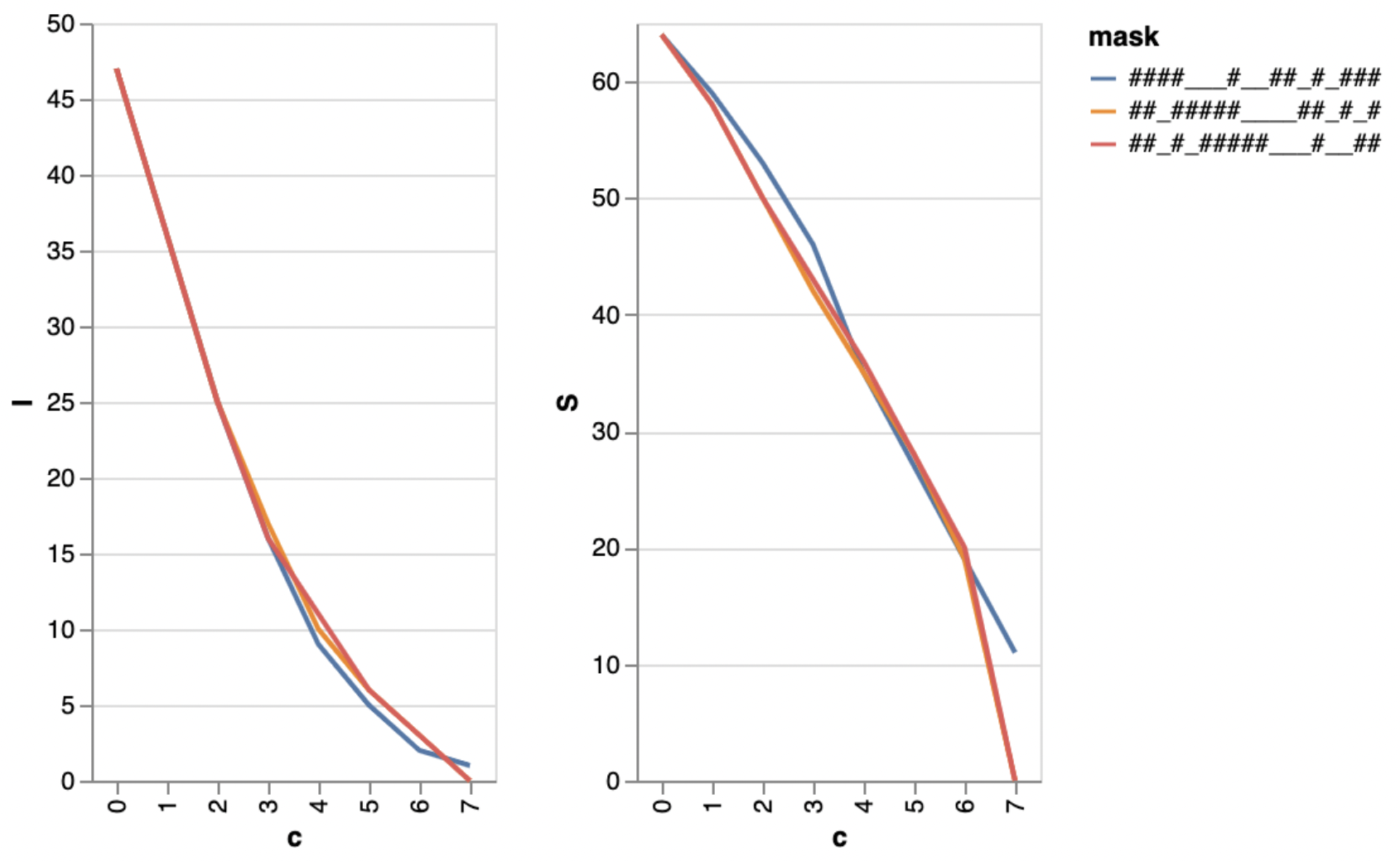
 Investigators: Bianca Stöcker, Till Hartmann, Johannes Köster & Sven Rahmann
Investigators: Bianca Stöcker, Till Hartmann, Johannes Köster & Sven Rahmann
Collaborators: Collaborative Research Center SFB 876
Funding: DFG SFB 876/3, project C1
Summary: We build and optimise models for clinically relevant decisions in oncology by selecting features from high-dimensional feature spaces, extracted from raw data created on different molecular platforms.
(Details)
Methylation analysis of whole genomes and targeted regions
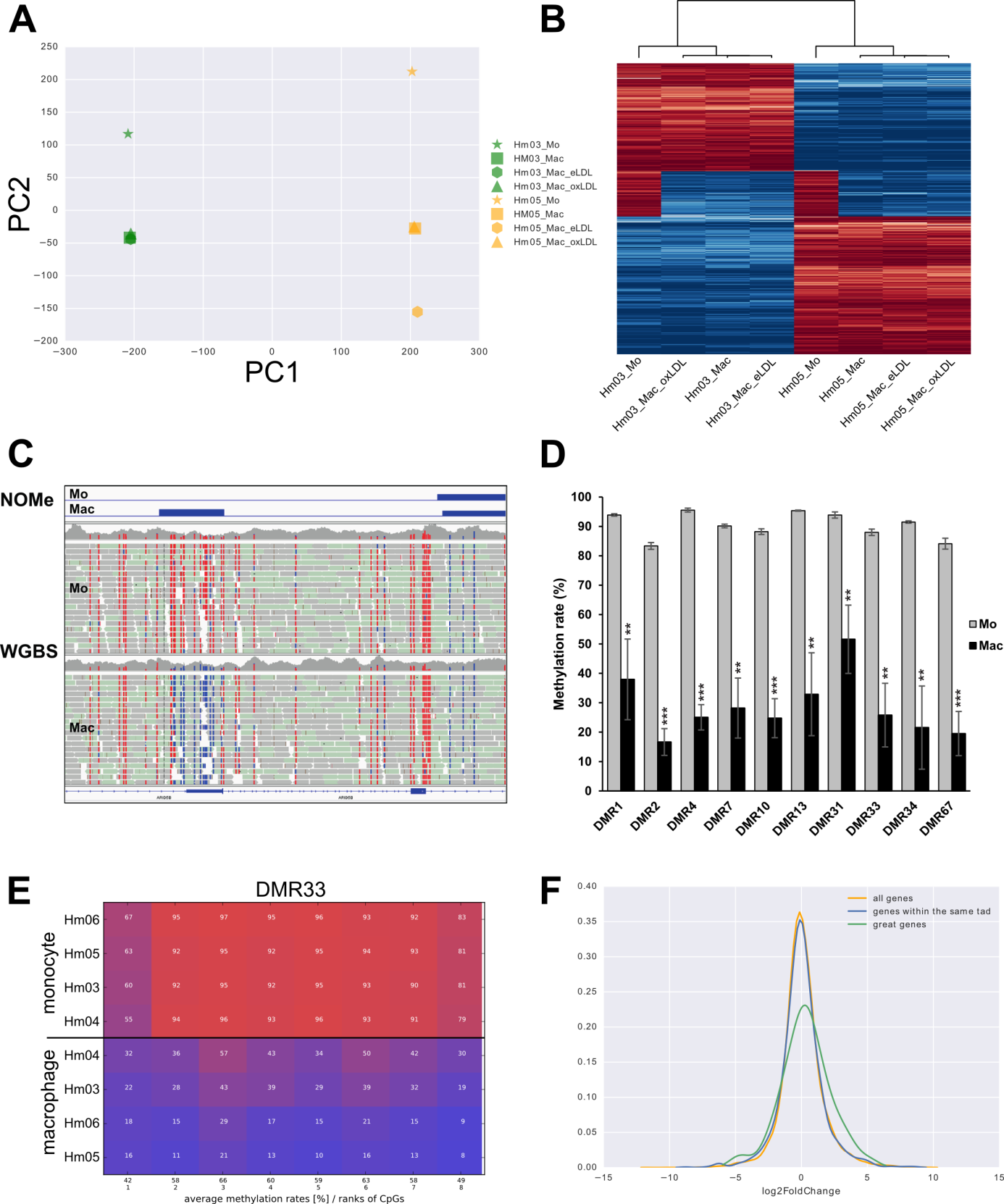 Investigators: Christopher Schröder, Marcel Bargull, Michelle Zechner & Sven Rahmann
Investigators: Christopher Schröder, Marcel Bargull, Michelle Zechner & Sven Rahmann
Collaborators: Bernhard Horsthemke (Human Genetics); formerly the DEEP consortium
Funding: internal; formerly BMBF (DEEP project)
Summary: We develop novel methods to analyze methylation levels across whole genomes from bisfulite sequencing data (WGBS) and on targeted regions (amplicons).
(Details)
Software: CAMEL
Mathematics of diversity
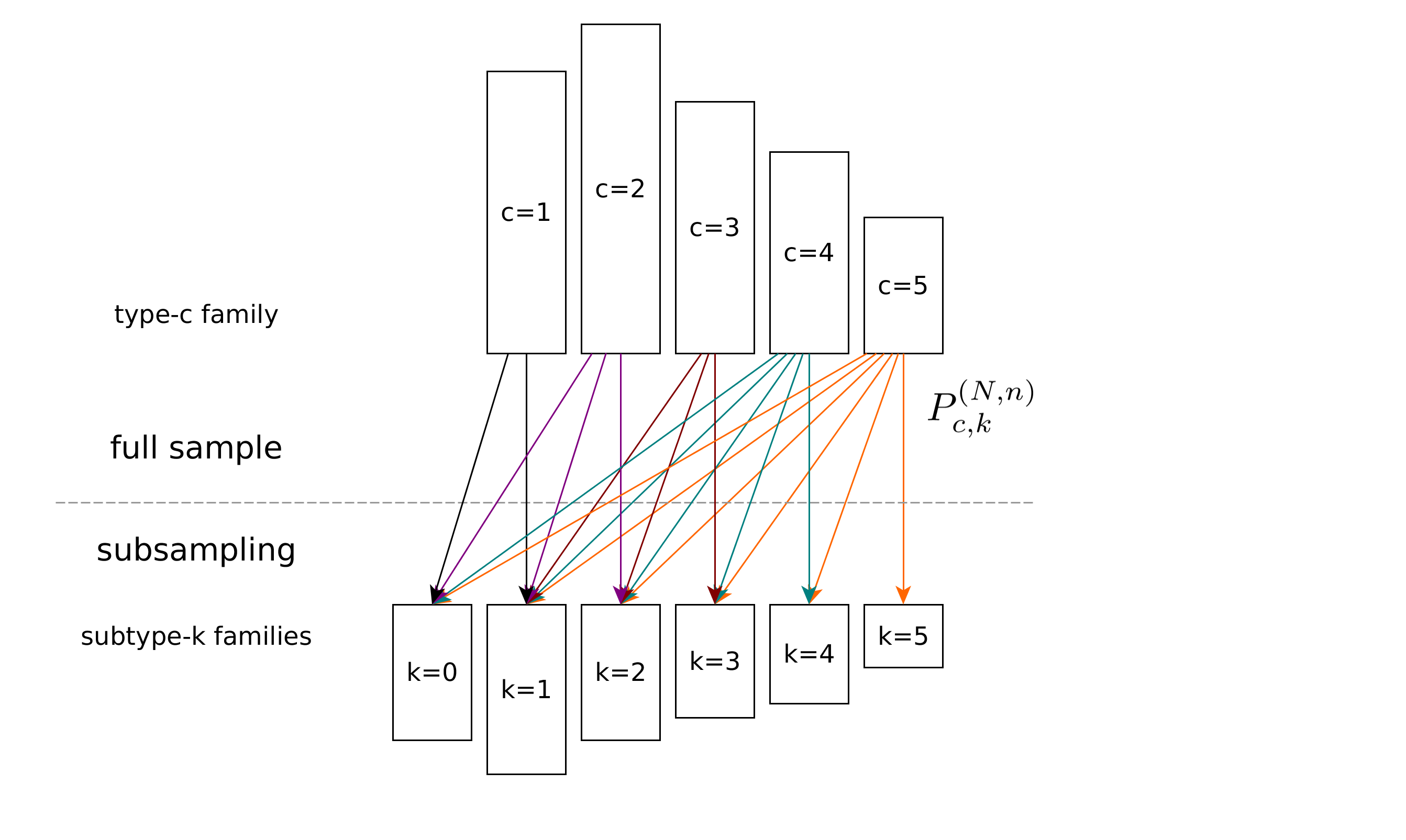 Investigators: Sven Rahmann
Investigators: Sven Rahmann
Funding: internal
Summary: We investigate the diversity of large populations when only a small subsample can be observed. Our methods come from linear and convex optimization.
(Details)
Software: dupre
Protein hypernetworks and protein complex similarity
 Investigators: Bianca Stöcker, Johannes Köster & Sven Rahmann
Investigators: Bianca Stöcker, Johannes Köster & Sven Rahmann
Collaborators: Eli Zamir
Funding: internal
Summary: We present an approach for endowing protein interaction networks with interaction dependencies using propositional logic, thereby constraining protein complex formation. We also developed a topology-derived similarity measure between protein complexes.
(Details)
Exome analysis with EAGLE
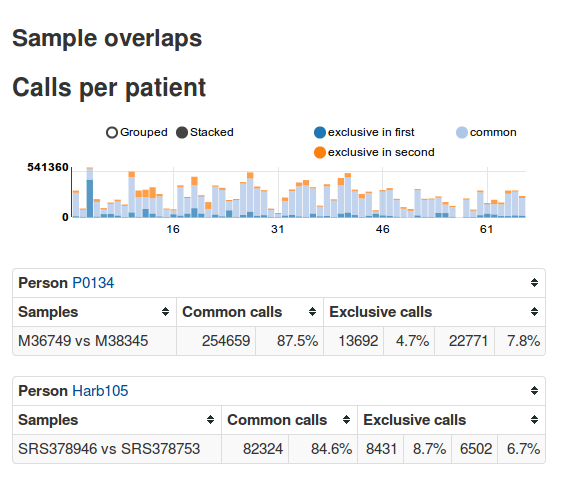 Investigators: Christopher Schröder, Felix Mölder & Sven Rahmann
Investigators: Christopher Schröder, Felix Mölder & Sven Rahmann
Collaborators: Institute for Human Genetics, University Hospital Essen
Funding: Mercator Research Center Ruhr (2010-2012); now internal
Summary: EAGLE is an exome-sequencing pipeline with a web frontend. It automates most stepsfrom FASTQ files to variant calls, stores the calls and metadata about patients, samples, etc. in a database and allows interactive analysis via a web frontend.
(Details)
Software: EAGLE
Metatranscriptomics
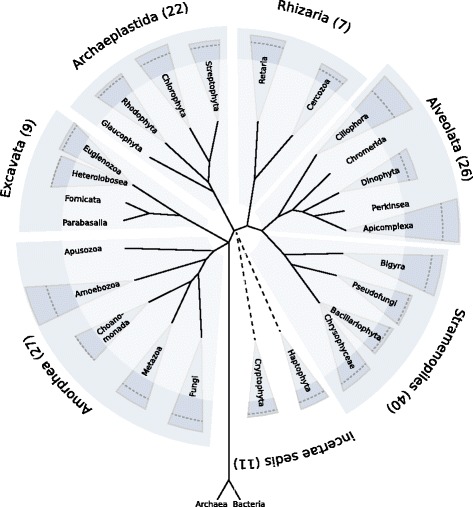 Investigators: Daniela Beisser & Sven Rahmann
Investigators: Daniela Beisser & Sven Rahmann
Collaborators: Jens Boenigk (biodiversity chair), Essen
Funding: internal
Summary: We developed a reference database, workflow and tool to assign reads from eukaryotic metatranscriptome experiments to both species and a functional class.
(Details)
Software: TaxMapper
Flexible until it snaps: Dynamics of genes & traits, densities & diversity in communities challenged by environmental change
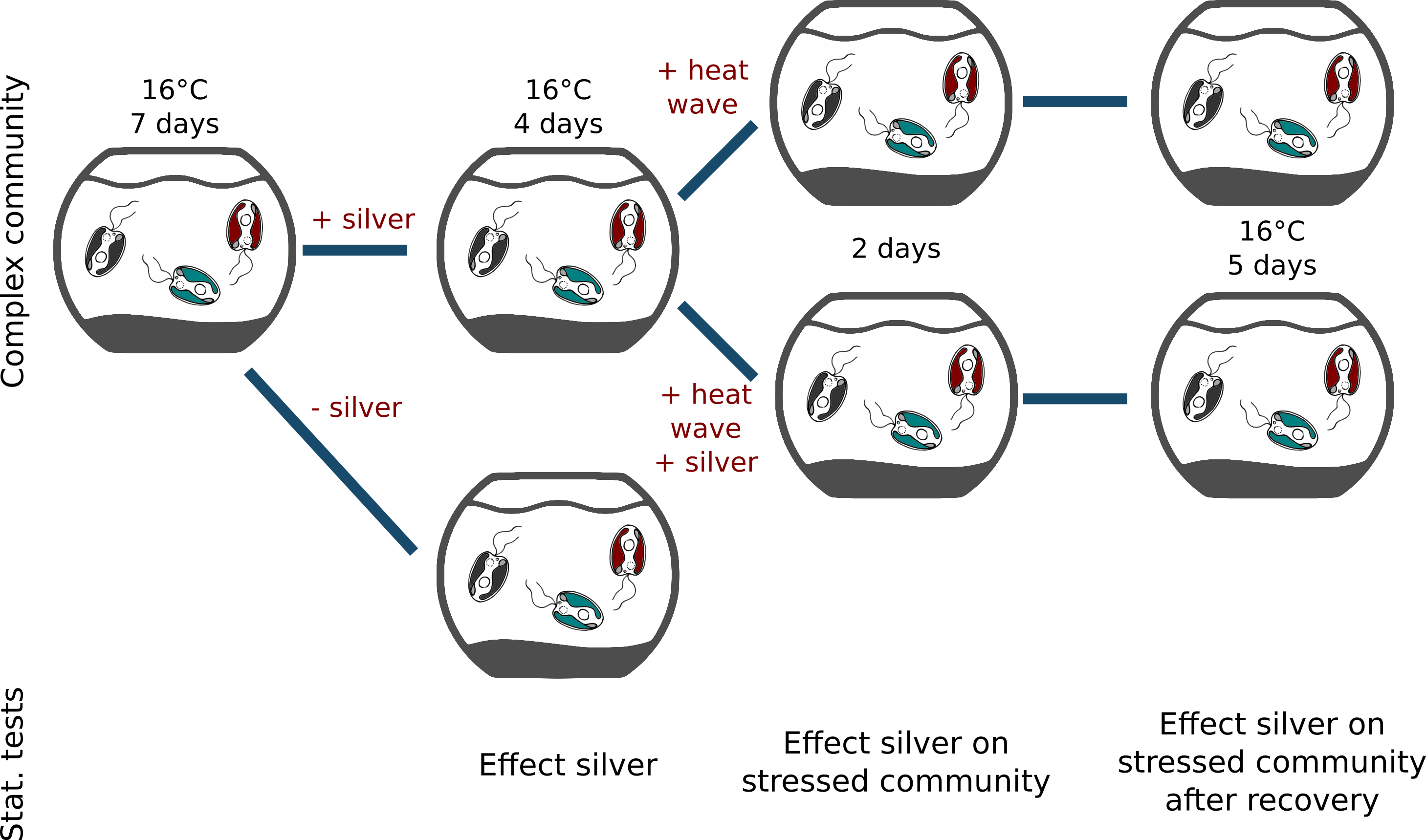 Investigators: Daniela Beisser & Sven Rahmann
Investigators: Daniela Beisser & Sven Rahmann
Collaborators: Jens Boenigk and Christina Bock (Biodiversity, Essen), Bernd Sures (Essen) and Matthijs Vos (Theoretical and Applied Biodiversity, Bochum)
Funding: DFG Priority Programme SPP 1704 "Flexibility matters: Interplay between trait diversity and ecological dynamics using aquatic communities as model systems" (Dynatrait)
Summary: We examine protists' dynamic responses to environmental stressors, specifically to ionic silver, and on the effect of such stressors to constrain the communities ability to dynamically respond to additional stressors such as heat waves.
(Details)
YeastScent - Volatile metabolites as quantitative proxy for metabolic network operation of Saccharomyces cerevisiae
 Investigators: Elias Kuthe & Sven Rahmann
Investigators: Elias Kuthe & Sven Rahmann
Collaborators: Lars Blank, Christoph Halbfeld, Birgitta E. Ebert (RWTH Aachen), Sebastian Engell, Sven Wegerhoff (TU Dortmund), Jörg Ingo Baumbach, Ann-Kathrin Sippel (Reutlingen), Jessica Kuhlmann (B&S Analytik GmbH, Dortmund), Michael Quantz, Erik Pollmann (Versuchsanstalt der Hefeindustrie e.V., Berlin)
Funding: BMBF project 031A301C
Summary: We aim to discover volatile organic compounds that precede the crabtree effect (switch to ethanol production) during yeast fermentation in order to optimize biomass yield.
(Details)
Probabilistic Arithmetic Automata
Investigators: Tobias Marschall & Sven Rahmann
Funding: internal
Summary: Probabilistic Arithmetic Automata are an extension of Deterministic Finite Automata (DFAs) and Hidden Markov Models (HMMs) to allow arithmetic or arbitrary binary operations. Exact state-value probability distributions can be efficiently computed with in this framework, and many applications in string algorithmics and computational biology can be modeled in this framework and treated in a unifying manner.
(Details)
Software: MoSDi
Subsequence combinatorics
Investigators: Sven Rahmann
Collaborators: Cees Elzinga & Hui Wang
Funding: internal
Summary: We have investigated several combinatorial problems (enumeration problems) related to subsequences within a sequence.
(Details)
Software: SubSeqComb