Bioconda
 Description: a distribution of bioinformatics software realized as a channel for the versatile package manager Conda; contains thousands of software packages
(Homepage).
Description: a distribution of bioinformatics software realized as a channel for the versatile package manager Conda; contains thousands of software packages
(Homepage).
Author(s): Johannes Köster et al.




Description: a distribution of bioinformatics software realized as a channel for the versatile package manager Conda; contains thousands of software packages
(Homepage).
Author(s): Johannes Köster et al.
 Description: a fast, memory frugal and accurate tool to remove human contamination using a pangenomic spaced seed index
(Homepage).
Description: a fast, memory frugal and accurate tool to remove human contamination using a pangenomic spaced seed index
(Homepage).
Author(s): Jens Zentgraf, Johanna Elena Schmitz & Sven Rahmann
 Description: a hidden markov model for chromatin state discovery with flexible emission and duration modeling
(Homepage).
Description: a hidden markov model for chromatin state discovery with flexible emission and duration modeling
(Homepage).
Author(s): Johanna Elena Schmitz, Nihit Aggarwal et al.
 Description: a tool for counting gapped k-mers
(Homepage).
Description: a tool for counting gapped k-mers
(Homepage).
Author(s): Jens Zentgraf & Sven Rahmann
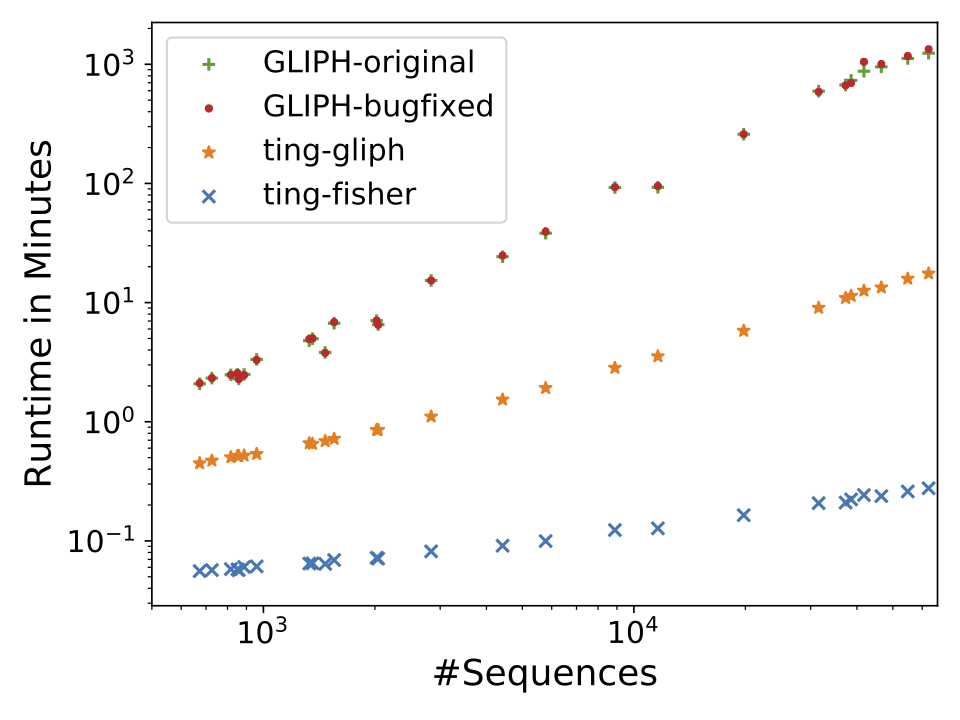 Description: a tool for clustering large scale T cell receptor repertoires by antigen-specificity
(Homepage).
Description: a tool for clustering large scale T cell receptor repertoires by antigen-specificity
(Homepage).
Author(s): Felix Mölder & Sven Rahmann
 Description: an ultrafast lightweight and accurate tool for xenograft sorting, using 3-way bucketed Cuckoo hashing
(Homepage).
Description: an ultrafast lightweight and accurate tool for xenograft sorting, using 3-way bucketed Cuckoo hashing
(Homepage).
Author(s): Jens Zentgraf & Sven Rahmann
 Description: two efficient fused lasso solvers for tree graphs; an implementation of the SEA 2020 paper by Elias Kuthe and Sven Rahmann
(Homepage).
Description: two efficient fused lasso solvers for tree graphs; an implementation of the SEA 2020 paper by Elias Kuthe and Sven Rahmann
(Homepage).
Author(s): Elias Kuthe
 Description: a tool to calculate a cost-optimal assignment of elements in multi-way bucketed Cuckoo hash tables; this provides an implementation of the method of the ALENEX 2020 paper by Jens Zentgraf, Henning Timm and Sven Rahmann.
(Homepage).
Description: a tool to calculate a cost-optimal assignment of elements in multi-way bucketed Cuckoo hash tables; this provides an implementation of the method of the ALENEX 2020 paper by Jens Zentgraf, Henning Timm and Sven Rahmann.
(Homepage).
Author(s): Jens Zentgraf & Sven Rahmann
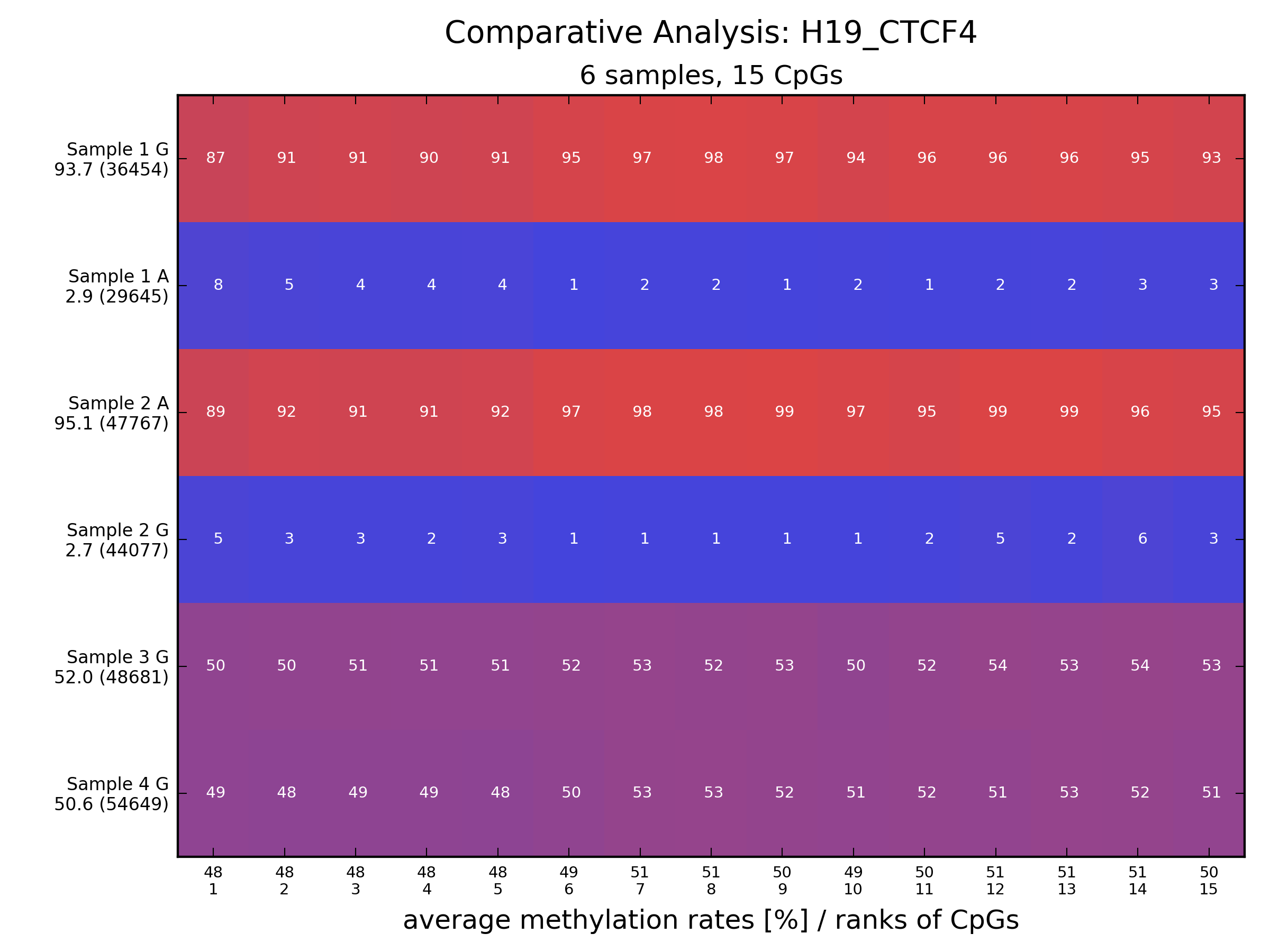 Description: an analysis tool for amplicon bisulfite sequencing that features automatic allele sorting; can also analyze NOMe-seq data
(Homepage).
Description: an analysis tool for amplicon bisulfite sequencing that features automatic allele sorting; can also analyze NOMe-seq data
(Homepage).
Author(s): Marcel Bargull, Sven Rahmann
 Description: A simulation tool for ddRAD-seq datasets that can produce a detailed and annotated ground truth for the evaluation of analysis pipelines and software.
(Homepage).
Description: A simulation tool for ddRAD-seq datasets that can produce a detailed and annotated ground truth for the evaluation of analysis pipelines and software.
(Homepage).
Author(s): Henning Timm & Sven Rahmann
 Description: a database, workflow and an analysis tool for reliably mapping sequenced datasets to a provided microeukaryotic reference
(Homepage).
Description: a database, workflow and an analysis tool for reliably mapping sequenced datasets to a provided microeukaryotic reference
(Homepage).
Research project: Metatranscriptomics
Author(s): Daniela Beisser, Henning Timm & Sven Rahmann
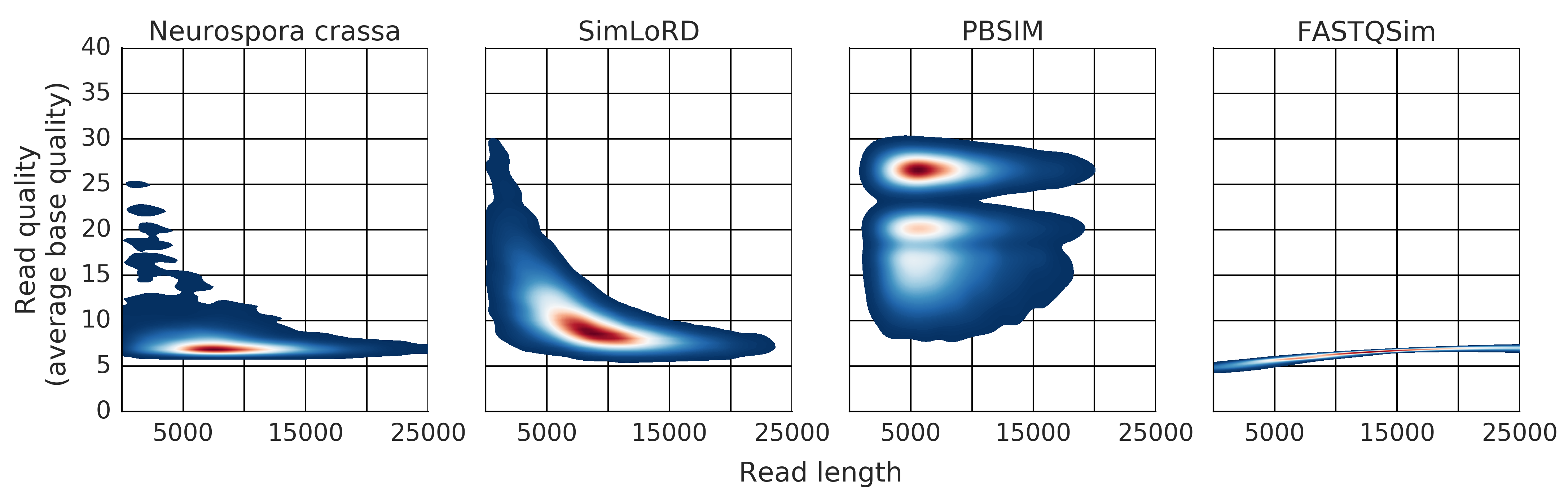 Description: a read simulator for third generation sequencing reads focused on the Pacific Biosciences SMRT error model
(Homepage).
Description: a read simulator for third generation sequencing reads focused on the Pacific Biosciences SMRT error model
(Homepage).
Author(s): Bianca Stöcker, Johannes Köster & Sven Rahmann
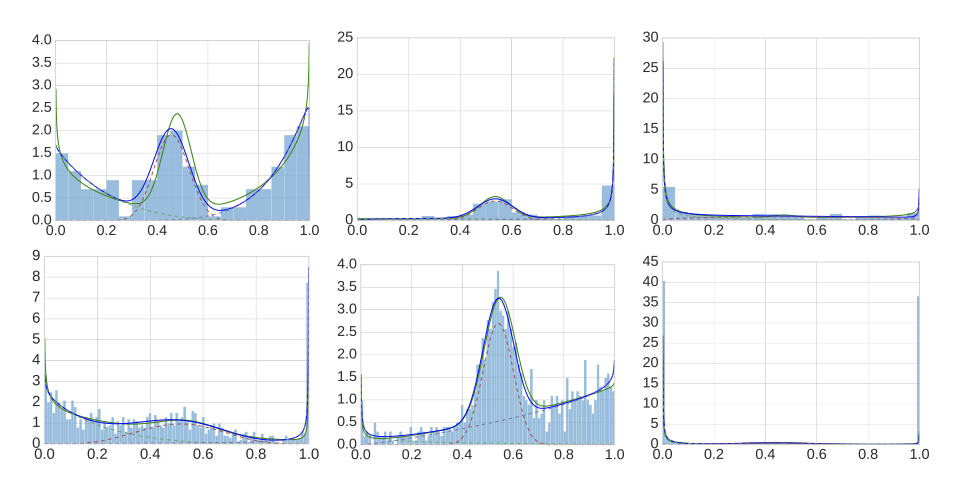 Description: a tool that robustly estimates a mixture of one-dimensional beta distributions to given datapoints in the interval [0,1]; motivated by the need to describe methylation level distributions.
(Homepage).
Description: a tool that robustly estimates a mixture of one-dimensional beta distributions to given datapoints in the interval [0,1]; motivated by the need to describe methylation level distributions.
(Homepage).
Author(s): Christopher Schröder & Sven Rahmann
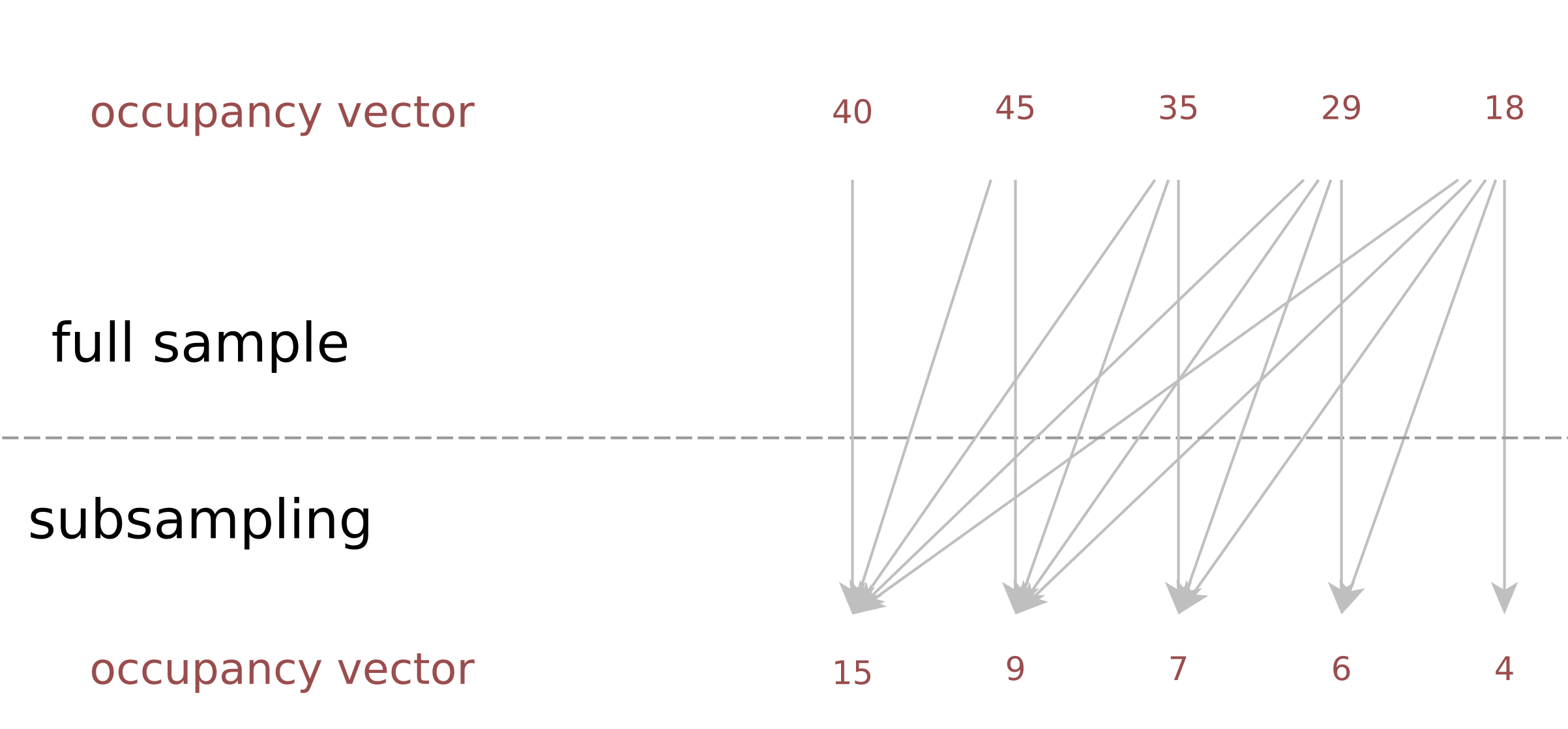 Description: a tool to estimate the duplicate rate of a sequencing library at an arbitrarysequencing depth, when the occupancy vector of a (small) subsample is known; useful for deciding decide which coverage to aim for, weighing new discoveries vsersus cost.
(Homepage).
Description: a tool to estimate the duplicate rate of a sequencing library at an arbitrarysequencing depth, when the occupancy vector of a (small) subsample is known; useful for deciding decide which coverage to aim for, weighing new discoveries vsersus cost.
(Homepage).
Research project: Diversity
Author(s): Christopher Schröder & Sven Rahmann
Description: methods for (DNA) motif statistics, e.g. to compute the exact occurrence count distribution of a motif, exact motif discovery, extraction of motifs with provably optimal p-value, analysis of pattern matching algorithms (to compute, for given algorithm and pattern, the exact distribution of the number of character accesses caused by searching a random text).
(Homepage).
Research project: Paas
Author(s): Tobias Marschall
Description: a web based frontend for argparse that allows launching and running python programs through a web browser.
(Homepage).
Author(s): Christoph Stahl
Description: a collection of fully automated peak extraction methods for MCC/IMS datasets, provided as a modular extensible framework backed by an open source implementation.
(Homepage).
Author(s): Marianna D'Addario, Dominik Kopczynski & Sven Rahmann
Description: a proof-of-concept implementation of flowgram-string alignment as described in our GCB 2013 paper “Aligning Flowgrams to DNA Sequences”
(Homepage).
Author(s): Marcel Martin & Sven Rahmann
Description: command line utility that scans a text or tex input file for patterns that indicate bad writing style (e.g., according to Strunk and White's classic "The Elements of Style") and reports occurrences of these, together with suggestions for improvement. Beta version. Retired.
(Homepage).
Author(s): Sven Rahmann
Description: Java classes for subsequence combinatorics. The Java class SubsequenceCombinatorics implements the algorithms described in our research project on subsequence combinatorics. The Java class SubsequenceCombinatoricsTest is a JUnit based test and at the same time demonstrates the use of the routines in SubsequenceCombinatorics.java. Documentation in html form is also included in the archive. In order to make the classes work on your system, you may have to change its package declaration.
(Homepage).
Research project: Subsequencecombinatorics
Author(s): Sven Rahmann
Algorithmic Bioinformatics, SIC, Saarland University | Privacy notice | Legal notice